Josep E. Esquerda Colell
Catedràtic Emèrit de la Facultat de Medicina (Universitat de Lleida)
La neurona és la unitat bàsica de funcionament del sistema nerviós, tal com fou formulat per Ramón y Cajal ja fa més de 100 anys. Cada neurona té una singularitat morfològica i funcional, que depèn de la seva articulació amb altres neurones de les quals rep milers d’influències mitjançant els contactes sinàptics. Podem entendre les sinapsis com les unitats bàsiques de la comunicació interneuronal, de les quals n’hi ha una gran diversitat de tipus pel que fa al seu sistema de funcionament basat, essencialment, en neurotransmissors químics; cada neurona rep inputs sinàptics aferents a través del seu soma i arbre dendrític; la lectura resultant de la integració temporal i espacial de tots aquests inputs determina la generació o no, d’una descàrrega en forma de potencial d’acció propagat per l’axó; això dibuixarà el patró binari de sortida de senyals en una neurona i que, al seu temps, acabarà amb l’activació de tots els terminals presinàptics que connecten dita neurona amb les altres amb les quals forma circuït.
Hem de considerar també, que a més de la comunicació sinàptica de caràcter ràpid i amb conseqüències de canvis elèctrics en el sentit de despolarització (excitació) o hiperpolarització (inhibició), també en el últims anys hem aprés que en la comunicació sinàptica es donen també fenòmens a llarg termini, que condicionen canvis plàstics en la cèl·lula postsinàptica vinculats a modificacions postraduccional de proteïnes, inducció de factors de transcripció reguladors de l’expressió gènica i altres fenòmens de caràcter més permanent. Aquests, són menys coneguts i estan relacionats amb fenòmens com la memòria i l’aprenentatge, la estabilitat sinàptica o la supervivència neuronal entre altres. A més dels neurotransmissors clàssics, hem aprés que també hi ha un nombre molt important i divers de factors de naturalesa peptídica amb funcions molt variades relacionades, per exemple, amb la supervivència i diferenciació neuronal, plasticitat sinàptica, i moltes altres. Tot això fa palesa la enorme complexitat de la comunicació interneuronal.
El nombre de sinapsis en un cervell és enorme, d’una magnitud astronòmica eixordant. El premi Nobel Gerard Edelman n’ha fet una estimació: calcula que en el còrtex hi ha 1000 X1012 sinapsis; per facilitar la comprensió del significat d’aquesta xifra, Edelman explica que, contades a una velocitat d’una per segon, es tardaria 32 X106 anys en finalitzar el recompte. Les possibles combinacions que es poden formar donen una xifra de 10 seguit de 106 zeros (el número de partícules positives en tot l’univers és de 10 segut de 80 zeros).
Les neurones són cèl·lules terminalment diferenciades, es a dir, que han perdut totalment i de forma irreversible la seva capacitat per reproduir-se. Per tant, qualsevol pèrdua neuronal en l’adult es irreparable. És el preu que s’ha de pagar per garantir l’estabilitat de funcions tant genuïnament humanes com la memòria, l’aprenentatge, el llenguatge, l’autoconsciència i els fenòmens emocionals; es a dir, l’activitat mental en el seu conjunt. La demostració de l’existència d’una neurogènesi postnatal en rosegadors, particularment en el cervell olfactiu i en l’hipocamp, resta encara ara controvertida en la seva vessant humana. En qualsevol cas, la seva significació en termes de reparació de la lesió neurològica és, probablement, escassa o nul·la.
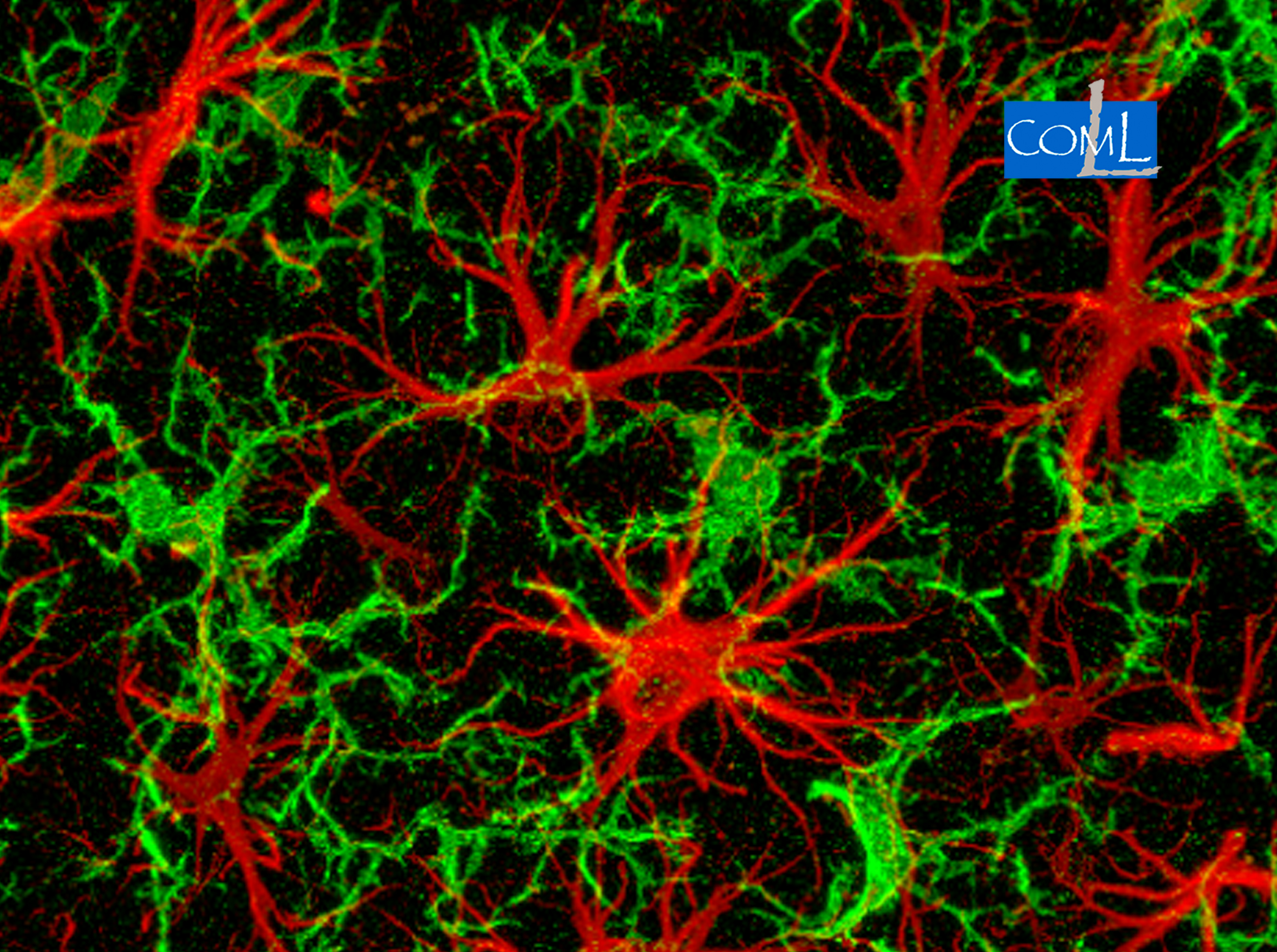
No podem deixar de fer referència en aquest petitíssim repàs introductori a la glia, constituïda bàsicament pels astròcits, la micròglia i la oligodendròglia en el SNC. Aquestes cèl·lules juguen un paper fonamental en el funcionament normal de les neurones i també tenen un gran protagonisme en tota patologia neurològica. Cal recordar el nom de Pío del Río Hortega ben lligat a la descoberta de la micròglia i de l’oligodendroglia. També comencem a conèixer alguns dels complexos viaranys de la comunicació neurona glia i a evidenciar-ne la seva gran importància neuropatològica.
En les malalties neurodegeneratives, es produeix primer una disfunció i després una pèrdua progressiva de neurones que afecta, de manera relativament selectiva, a determinats territoris del sistema nerviós central. La conseqüència òbvia es la pèrdua indeturable de les funcions associades als sistemes afectats tal com s’observa en la malaltia d’Alzheimer, de Parkinson, en l’esclerosi lateral amiotròfica (ELA), per exemple. A causa d’aquests processos, milions de persones en tot el món sofreixen dures malalties orfes de tractaments. L’augment de l’esperança de vida degut a l’esplendorós desenvolupament de la Medicina durant el segle XX, que ha permès curar o controlar altres patologies, ha comportat, per altra banda, un increment de la morbilitat i mortalitat per les malalties neurodegeneratives. Així doncs, la troballa de teràpies efectives és un dels reptes més importants que ha d’afrontar la Medicina del segle XXI. El problema es realment difícil, ja que la seva arrel s’endinsa cap a territoris d’altíssima complexitat i en gran part desconeguts relatius a la organització i el funcionament del sistema nerviós.
Malgrat se’n podrien considerar moltes altres, en referirem bàsicament a les tres malalties abans mencionades, fent particular esment de l’ELA, donat que aquesta és l’objecte de la activitat de recerca del nostre grup. Hem de tenir present també que la malaltia d’Alzheimer, la de Parkinson, i l’esclerosi lateral amiotròfica (ELA o malaltia de Charcot), malgrat tinguin una simptomatologia clínica molt diferent des del punt de vista neurobiològic, tenen característiques molt comparables. Això implica que l’avenç en el coneixement i estratègies terapèutiques en qualsevol d’elles té, sens dubte, repercussió sobre les altres. Són entitats clinicopatològiques en les que la degeneració i la pèrdua neuronal va acompanyada de dipòsits de proteïnes mal plegades, com la beta-amiloide i proteïnes citoesquelètiques en el cas de l’Alzheimer o de la α-sinucleïna en el Parkinson, amb efectes neurotòxics i d’inducció d’activació astro i microglial neuroinflamatòria que amplifica el dany. Podem considerar aquests processos com proteinopaties neuroinflamatòries. Això situa l’agregació patològica de proteïnes i la neuroinflamació com a dianes molt investigades per l’acció terapèutica. No obstant, segueix controvertit el paper fisiopatològic que juguen realment els dipòsits proteics en la malaltia, ja que la reducció de les plaques amiloides en la malaltia d’Alzheimer no sempre s’ha mostrat terapèuticament efectiva.
Un altra característica comuna d’aquestes malalties es l’existència de formes esporàdiques, que són les més comunes i formes familiars molt menys prevalents. En els últims anys s’han identificat un nombre apreciable de gens, la mutació dels quals causa alguna d’aquestes malaltias neurodegenerativas. En general, podem dir que el gen mutat codifica per una proteïna alterada amb tendència a formar agregats patològics que desencadenen neurotoxicitat i neuroinflamació. En els últims anys, la possibilitat de transferir alguns d’aquests gens alterats a animals transgènics ha donat lloc a la generació de models animals d’aquestes malalties que han estat essencials per al desenvolupament de la recerca en els últims anys. Gràcies a aquesta investigació preclínica, en el últims anys s’ha generat una quantitat notabilíssima de nou coneixement i ens ha permès realitzar assajos terapèutics experimentals que són imprescindibles per al plantejament d’assajos clínics ulteriors; malauradament, això no ha cristal·litzat, encara, en una descoberta significativa i d’aplicabilitat en clínica humana.
Pel que fa a l’ELA, aquesta malaltia fou descrita per un dels pares de la Neurologia moderna, Jean-Martin Charcot, que treballava a l’Hôpital de la Salpêtrière a Paris, encara vigent, on desenvolupava el procediment anatomoclínic en l’estudi les malalties neurològiques. Així va definir l’ELA com una malaltia que cursa amb paràlisi progressiva i atròfia muscular per afectació del sistema motor voluntari, tant a nivell de la 1a neurona en el còrtex com de la 2a en la banya anterior de la medul·la espinal. La afectació del tracte cortico-espinal determina la desmielinització i l’esclerosi dels cordons laterals de la medul·la; d’aquí ve el nom d’ELA proposat per Charcot a les acaballes del segle XIX.
Una estratègia similar, molt moderna a l’època, la practicava a Frankfurt, Alois Alzheimer al definir la malaltia que porta el seu nom en la primera dècada del segle XX.
Pel que fa a l’ELA, és una malaltia de l’adult, amb una prevalença de entre 4-6 cassos per 100.000. El 90% dels casos apareix de forma esporàdica, sense que hi hagi cap component genètic obvi. En el 10 % de casos restants, l’ELA és una malaltia hereditària amb caràcter dominant. Des del punt de vista clínic, ambdós formes presenten característiques cardinals similars com la debilitat muscular progressiva, amb atròfia (signe d’afectació de la 2a neurona motora) i espasticitat (signe d’afectació de la 1a neurona motora); la denervació progressiva dels músculs respiratoris és un element determinant del seu curs fatal entre 1 i 5 anys després del diagnòstic. En general, l’intel·lecte i les capacitats cognitives estan preservades, però també hi ha cassos associats a demència frontotemporal, particularment en aquelles formes familiars degudes a la mutació de gen C9orf72 o del que codifica per la proteïna TDP-43.
La primera alteració genètica causant d’ELA familiar que es va identificar és la del gen que codifica per la proteïna superòxid dismutasa 1 (SOD1), la qual cosa succeí l’any 1993 i, poc temps després, un gen mutant de SOD1 causant d’ELA familiar humana, va ser insertat en un ratolí transgènic, el qual va desenvolupar un fenotip comparable a l’ELA humana. Va ser el primer model animal de la malaltia i segueix sent, fins al nostres dies, el més treballat en la recerca preclínica sobre l’ELA. Malgrat que aquest model representaria menys d’un 10% de les formes familiars d’ELA, es considera que, des del el punt de vista de mecanismes fisiopatològics y de dianes terapèutiques a assajar, tindria prou trets en comú amb les altres formes d’ELA. La patogenicitat de la SOD1 mutada no està relacionada amb el dèficit de la seva activitat enzimàtica dismutàsica en la defensa contra l’estrès oxidatiu; més aviat està relaciona amb un guany de funció tòxica de naturalesa desconeguda. Les formes mutades i neurotòxiques de SOD1 tenen tendència a formar agregats patològics i també d’interferir de manera anòmala en una diversitat de processos cel·lulars essencials per al funcionalment neuronal. Entre aquest, són particularment destacables les alteracions mitocondrials del reticle endoplàsmic i control de qualitat en la síntesi de proteïnes, del transport i metabolisme del RNA, del transport axoplàsmic i moltes altres. També la SOD1 alterada determina activació neurotòxica de la astroglia i de la micròglia adjacent a les motoneurones afectades. També la astroglia es defectiva en l’expressió de transportador de glutamat que n’elimina el seu excés en l’espai extracel·lular; això provoca una estimulació exagerada i excitotòxica dels receptors de glutamat a la motoneurona. De fet, el riluzol, fins ara fa poc l’únic fàrmac empleat en tractament de l’ELA (amb eficàcia molt limitada), té efectes anti-glutamatèrgics. Tots el mecanismes anteriorment esmentats constitueixen possibles dianes per a l’acció terapèutica que s’han investigat en un gran nombre d’estudis experimentals sense que cap d’ells hagi traspassat a la clínica, excepte la molècula antioxidant edaravone, aprovada per a la seva administració en pacients d’ELA, primer al Japó i després a EEUU amb efectes terapèutics realment molt modestos i, en qualsevol cas, controvertits. Tot això ens situa en la necessitat d’implementar encara molt més la recerca des dels seus nivells més bàsics fins a aquells més translacionals.
En els últims anys, s’han identificat altres gens implicats en les formes familiars d’ELA, com per exemple, c9orf72, TARDBP, que codifica per TDP-43, FUS, Alsin, ErbB4 i altres. En relació als mecanismes patogènics implicats en la forma esporàdica, el grau de coneixement és encara més escàs si el comparem amb les formes familiars. Tenim dades recents molt xocants, que indiquen que la SOD1 salvatge (WT) pot adquirir propietats neurotòxiques per modificacions post-traduccionals adquirides. Això posaria de manifest mecanismes patogènics comuns entre algunes formes familiars d’ELA i les formes esporàdiques en els que la SOD1 ocuparia un rol central. Això apunta cap a aquesta molècula com un nova diana terapèutica d’ampli espectre en diverses formes clíniques d’ELA i atorga un interès renovat als models d’animals transgènics basats en SOD1.
Un aspecte relativament emergent en la recerca sobre l’ELA i en altres malalties neurodegeneratives proteinopàtiques és l’existència d’un mecanisme de propagació de la malaltia similar a l’existent en les malalties priòniques. Per exemple, en malalties com l’Alzheimer, el Parkinson, la demència frontotemporal o proteinopaties amb repeticions de poliglutamina (com la malaltía de Hungtington) es produeix una acumulació d’agregats de proteïnes com tau, β-amiloide, α-sinucleïna o hungtintina, que podria propagar-se cèl·lula a cèl·lula; seria com una espècie de llavor, comunicant i amplificant el mal plegament a les formes encara no alterades de la proteïna. Un exemple recent d’aquest mecanisme es la demostració, en una publicació recent (un equip de la Johns Hopkins University School of Medicine a Baltimore), que la injecció de fibril·les patològiques de α-sinucleïna a la paret del tub digestiu a nivell duodenal o pilòric, determina la seva propagació i viatge fins al cervell on provoca degeneració de neurones dopaminèrgiques i simptomatologia clínica de Parkinson; aquestes alteracions s’eviten per la secció del nervi vago, demostrant així el transport i propagació transneuronal de la proteïna alterada. Una vegada reconeguda la capacitat d’algunes proteïnes i agregades (especialment en les formes en fulla β i amiloides), generadores de patologia neurodegenerativa, per propagar-se de forma intermolecular i transcel·lular, s’ha introduït el terme de “prionoide” per referir-se a aquesta propietat i, a la vegada, diferenciar-lo del mot “prió” que comporta, addicionalment, la demostració de la seva transmissibilitat entre individus, equiparable a allò que succeeix amb qualsevol altre agent infecciós. Això a més del seu impacte notable en la concepció intel·lectual de la malaltia neurodegenerativa, té també importants conseqüències en el desenvolupament de futures teràpies.
Pel que fa a la SOD1, hi ha dades que suggereixen que el mecanisme “prionoide” pot operar tant en l’ELA familiar com a l’esporàdica. La proteïna implicada en ELA, TDP-43 també tindria aquesta propietat. Una manera de frenar aquesta propagació intercel·lular podria ser segrestar la proteïna en l’espai extracel·lular mitjançant anticossos específics. Aquests anticossos també podrien ajudar a dissoldre els agregats proteics i modular l’activitat microglial com s’ha vist en la malaltia d’Alzheimer administrant, per exemple, l’aducanumab, un anticòs monoclonal d’origen humà actualment en procés de possible aprovació per una administració més àmplia. Seguint una estratègia similar, la mateixa companyia (Neurimmune AG; Zurich) ha aïllat anticossos contra formes mal plegades de SOD1 a partir de la clonació de limfòcits humans d’individus vells (centenaris). Aquests anticossos tenen un efecte notablement beneficiós sobre la evolució i supervivència de ratolins amb ELA. Aquesta podria ser una nova estratègia de teràpia que, sens dubte, es desenvoluparà en els propers anys i que probablement formarà part d’un tractament combinat amb altres fàrmacs actuant sobre altres de les múltiples dianes moleculars que la recerca bàsica posa de manifest.
En quant a la teràpia cel·lular, és del tot ingenu pensar en una teràpia substitutiva dels elements neuronals perduts per neurodegeneració. S’han fet molts experiments en aquest sentit, particularment en aquelles patologies en les que la pèrdua neuronal esta relativament confinada en l’espai com és en el cas de la malaltia de Parkinson. Els processos de propagació prion-like limiten aquesta estratègia, ja que s’ha vist que les cèl·lules dopaminèrgiques normals trasplantades a humans desenvolupen amb el temps inclusions proteiques de tipus Lewy, indicant que han adquirit per contagi la patologia de l’hoste. Sí, en canvi, es pot considerar plausible la introducció de cèl·lules modificades adequadament amb intenció d’alterar positivament un entorn neuronal desfavorable, però tot això requereix encara molta recerca experimental.
En el nostre grup de recerca al Departament de Medicina Experimental i a l’IRBLLEIDA, es treballa en models animals d’ELA i també d’una malaltia de la motoneurona que afecta a joves infants com és l’atròfia muscular espinal (AME). Des del 2005, disposem d’una colònia de ratolins transgènics SOD1 i també vam ser pioners a l’Estat espanyol amb la disposició de models animals d’AME. La nostra aproximació a aquestes malalties es sota l’òptica de l’anàlisi als nivells cel·lular i molecular: es tracta de posar de manifest dianes per a possibles noves estratègies de teràpia. També aprofitem la disponibilitat d’aquests models in vivo per a la realització d’assajos preclínics. En els últims anys, la nostra tasca ha estat dirigida, més específicament, a l’anàlisi dels aferents sinàptics a la motoneurona i el seu paper en la patologia. Hem identificat nous components moleculars en les anomenades sinapsis de tipus C (pròpies de les motoneurones vulnerables a l’ELA) que indiquen mecanismes de senyalització amb possible rellevància patològica. Vinculat a l’alteració dels aferents sinàptics també hem i estem estudiant els processos d’activació astro- i microglial que son la base de la resposta neuroinflamatòria en la motoneurona lesionada. Els resultats d’aquesta investigació han estat publicats en revistes científiques d’impacte i difusió internacional, que es poden trobar fàcilment en les bases de dades de bibliografia biomèdica.